Polycystic kidney disease is a chronic, incurable illness estimated to affect between 1 in 500 and 1,000 people (Garvan Institute 2021; Better Health Channel 2017).
What is Polycystic Kidney Disease?
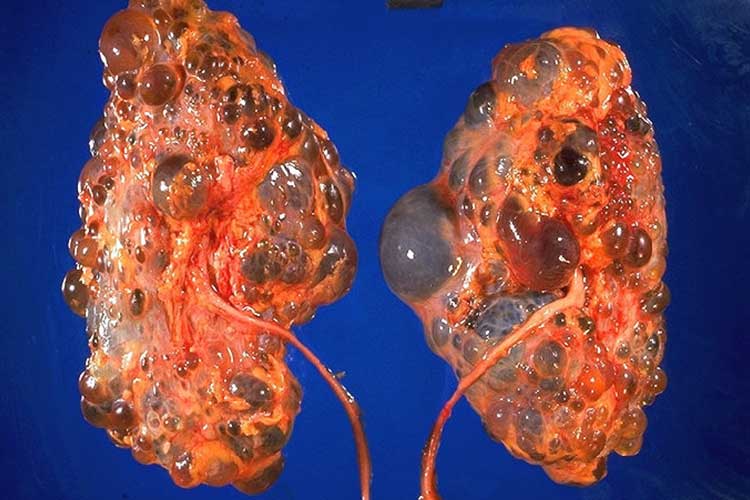
Polycystic kidney disease (PKD) is a predominantly genetic condition causing cysts (fluid-filled blisters) to grow on the kidneys (Better Health Channel 2017).
These cysts gradually enlarge the kidneys as they grow, causing healthy kidney tissue to be compressed. Eventually, this impairs kidney function and in some cases, leads to kidney failure (Better Health Channel 2017; Kidney Health Australia 2019).
Both kidneys are affected, but one might progress earlier than the other (Kidney Health Australia 2019).
As well as in the kidneys, cysts may also grow in other organs, including the liver, pancreas, spleen, ovaries, large bowel, heart and brain (NKF 2018).
Image credit to CDC/Dr. Edwin P. Ewing, Jr.
What Causes Polycystic Kidney Disease?
PKD occurs due to a mutation in the PKD1, PKD2 or PKHD1 gene (Better Health Channel 2017).
This mutation is believed to cause increased cell growth, which leads to the formation of cysts (Bennett et al. 2020).
In 90% of cases, the mutation is inherited, however, it’s also possible for PKD to occur in someone with no family history of the illness. This is thought to be caused by a spontaneous gene mutation or a different inheritance pattern (Garvan Institute 2021; Better Health Channel 2017).
There are two inheritance patterns of PKD:
Autosomal dominant (ADPKD)
Autosomal recessive (ARPKD).
(Kidney Health Australia 2019)
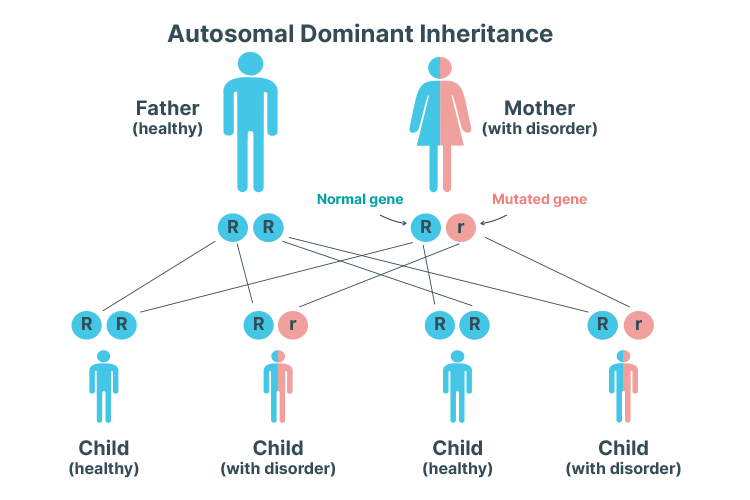
Autosomal Dominant PKD
Autosomal dominant inheritance pattern means that only one copy of the mutated gene is needed to cause PKD.
ADPKD is the most common type of PKD (Kidney Health Australia 2019).
In ADPKD, cysts begin to form during childhood but are initially microscopic and impossible to detect until later in life. Symptoms don’t typically develop until between 30 and 40 years of age (Kidney Health Australia 2019).
An autosomal dominant inheritance pattern means that only one copy of the mutated gene is needed to cause PKD. In other words, a child has a 50% chance of inheriting PKD if one of their parents has the mutated gene (Mayo Clinic 2020).
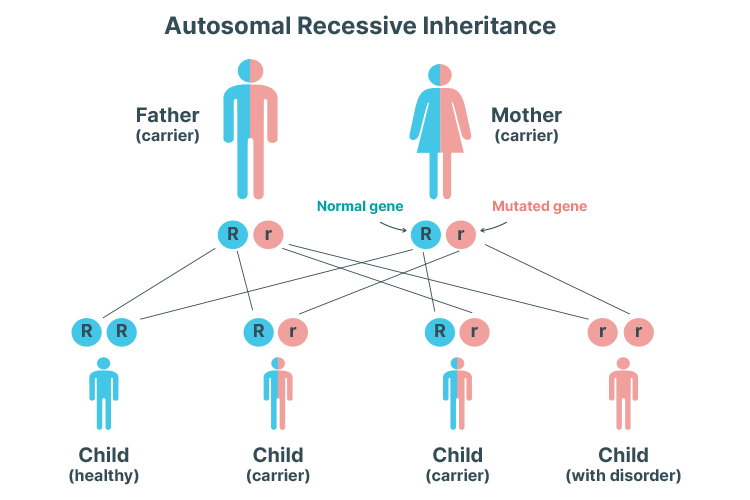
Autosomal Recessive PKD
It's only possible to inherit ARPKD if both parents are carriers of the mutated gene.
ARPKD is much less common. Unlike ARPKD, symptoms typically begin to develop during early childhood, or in some cases, as early as in the womb (Better Health Channel 2017).
ARPKD can cause kidney and/or liver problems later in the patient’s life (Kidney Health Australia 2019).
An autosomal recessive inheritance pattern means that two copies of the mutated gene are needed to cause PKD. In other words, the condition can only be inherited if both parents are carrying the mutated gene and the child inherits one mutated gene from each parent. One mutated gene is not enough to cause PKD alone - instead, the child will become an asymptomatic carrier. The likelihood of inheriting two copies of the mutated gene is 25% (Mayo Clinic 2020).
Symptoms of Polycystic Kidney Disease
Autosomal Dominant PKD
People with ADPKD don’t usually present with symptoms in early life, but between the ages of 30 and 40 (on average), they may begin to experience:
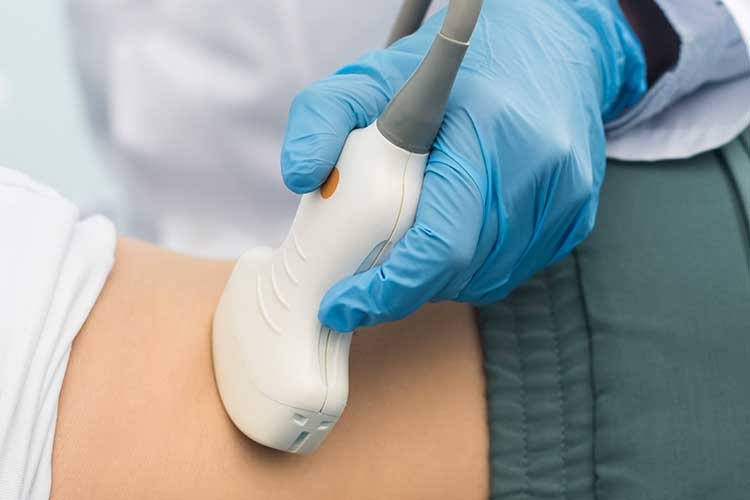
The most reliable, cost-effective and non-invasive method of detecting cysts is via an ultrasound.
Autosomal recessive PKD can usually be diagnosed early due to patients commonly presenting with severe symptoms at a young age (Kidney Health Australia 2019).
Autosomal dominant PKD, on the other hand, may go unnoticed for many years and is often detected during investigations for other health issues (e.g. urinary tract infection). Some people with ADPKD aren’t diagnosed until their kidneys begin to fail (Better Health Channel 2017).
Diagnosis of PKD will take into consideration the patient’s age, as well as any family history of the condition (Kidney Health Australia 2019).
The most reliable, cost-effective and non-invasive method of detecting cysts is via an ultrasound (NKF 2018).
If cysts are detected, further diagnostic tests might be performed, including:
Physical examination to detect hypertension or enlarged kidneys
Blood tests to check kidney function
Urine tests to assess for haematuria and proteinuria (protein in urine).
(Kidney Health Australia 2019)
In some cases, genetic testing might be used, but it isn’t a routine diagnostic method as it’s costly and isn’t always accurate (NKF 2018).
PKD will be diagnosed in at-risk people with a family history of the condition if:
Patient age
Number of cysts found on ultrasound
15-39 years
At least 3 (in total)
40-59 years
At least 2 in each kidney
< 60 years
At least 4 in each kidney
(Kidney Health Australia 2019)
Treatment for Polycystic Kidney Disease
There is no cure for PKD, but early detection and management can help to reduce the risk of complications (Better Health Channel 2017).
Typically, first-line treatment involves slowing the growth of cysts via lifestyle management and modification (e.g. regular exercise, smoking cessation, dietary changes), along with controlling blood pressure. In some cases, these are the only interventions needed (Kidney Health Australia 2019).
Other interventions may include:
Antihypertensive medicine to manage hypertension
Draining cysts to relieve pain
Fluids, analgesia, antibiotics and rest to treat haematuria
Antibiotic treatment for any urinary tract infections
Medicine (tolvaptan) to slow the progression of cysts (for adults with rapidly progressing ADPKD)
Dialysis or a kidney transplant to treat kidney failure
Psychological support to help manage feelings of sadness and anxiety associated with a PKD diagnosis
Avoiding contact sports if the kidneys, liver, spleen or abdomen are enlarged, as these organs could potentially be injured if hit.
(Kidney Health Australia 2019; Better Health Channel 2017)
Patients should also be advised to avoid taking non-steroidal anti-inflammatory drugs (NSAIDs) as they can potentially worsen kidney function (Better Health Channel 2017).
Test Your Knowledge
Question 1 of 3
Which type of polycystic kidney disease is easier to diagnose?
 New
New